New European regulation identifies brain stimulation devices approved for medical use

Understanding medical device regulations can be a struggle. Rapid technological development is often a headache in terms of regulations, and the recent developments in the brain stimulation field have not been any different. When discussing with healthcare professionals, we have often faced uncertainty about what devices can be used for patient treatment. The new regulation brings clarity to this question.
From May 26th onwards, the new European law regulating medical devices will be fully applicable after a transition period that started back in 2017 when the regulation was approved.
The European regulation 2017/745 on medical devices, or MDR (medical device regulation) for short, will apply to all medical devices that come to market after 26th of May 2021, and will replace the old Medical Device Directive (MDD).
While the previous directive could be applied to a national level by adapting the European indications to a national law, allowing each country to modify and adapt the directive, the new regulation must be adopted directly, overruling the ability of state members to adapt the legislation and forcing them to apply it unmodified.
What happens to the medical devices that where approved under the old regulation?
The new legislation takes into account the currently existing MDD devices, that is, devices that were certified for medical use under the old directive.
Like that, the MDR opens a possibility to keep old MDD devices in the market with a “legacy device status” for a few years provided that they have been brought to market before a notified body. The notified body that regulated the device must still exist and continue to monitor them, and no substantial changes can be done upon the device. If a substantial change is made, the device will lose the legacy device status and will to be certified again under the new regulation.
How does the new regulation affect tDCS devices?
A key change affecting the brain stimulation field is comes already on the article 1(2):
“This Regulation shall also apply, as from the date of application of common specifications adopted pursuant to Article 9, to the groups of products without an intended medical purpose that are listed in Annex XVI…”
Previously, a device could fall into medical device regulation depending on which kind of uses and applications their creators claim the device has. Taking advantage of that, some tDCS manufacturers did not claimed any therapeutical benefits nor applications for their products so they could launch them to the market without any kind of control.
However, with the MDR regulation, there’s a new annex establishing that certain products or devices without an intended medical purpose must be regulated. Among the list of products, the regulation will be applicable to “equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain”.
In practice, this means that all brain stimulation, like tDCS, tACS, tRNS, rTMS, etc, devices must comply with the medical device regulation. This is now unequivocal answer to the uncertainty. Only medical devices can be used for brain stimulation.
What happens then with non-regulated tDCS devices?
As brain stimulation devices that were launched to the market without approval of a notified body have not previously been certified by MDD, they cannot claim the status of legacy device. These devices must now comply with the MDR regulation or they will not be observant to EU regulations and therefore, cannot be sold on European markets.
Currently there are several tDCS devices that can be sold in EU markets, among which only a few can claim the legacy device status– one of them being Sooma Therapy. This renders the majority of the current devices’ illegal under the new regulation.
TDCS is the treatment method offered by Sooma for depression (Sooma Depression Therapy, indicated for Major Depressive Disorder) and chronic pain (Sooma Pain Therapy, indicated for Fibromyalgia and chronic neuropathic pain). By using Sooma devices, you ensure that you are performing patient treatment, should be it in the hospital or at patients’ home, with legal, regulated, tested and effective equipment that complies with the latest EU regulations.
How can a medical professional identify which devices to use?
At this point, there two ways in which one can differentiate those devices which have been approved for medical use from those which hasn’t:
- Declaration of conformity document. Every medical device must have an associated declaration of conformity document, which defines the regulations that the device complies with. The device manufacturer must be able to provide this document upon request. If it does not, then there is a high chance that the device has not been certified.
- Device label. The device’s label must show a CE mark on it, followed with a 4-digit number. This is the visual seal of approval of a notified body (a third-party auditor), which means that the device is an approved medical device.
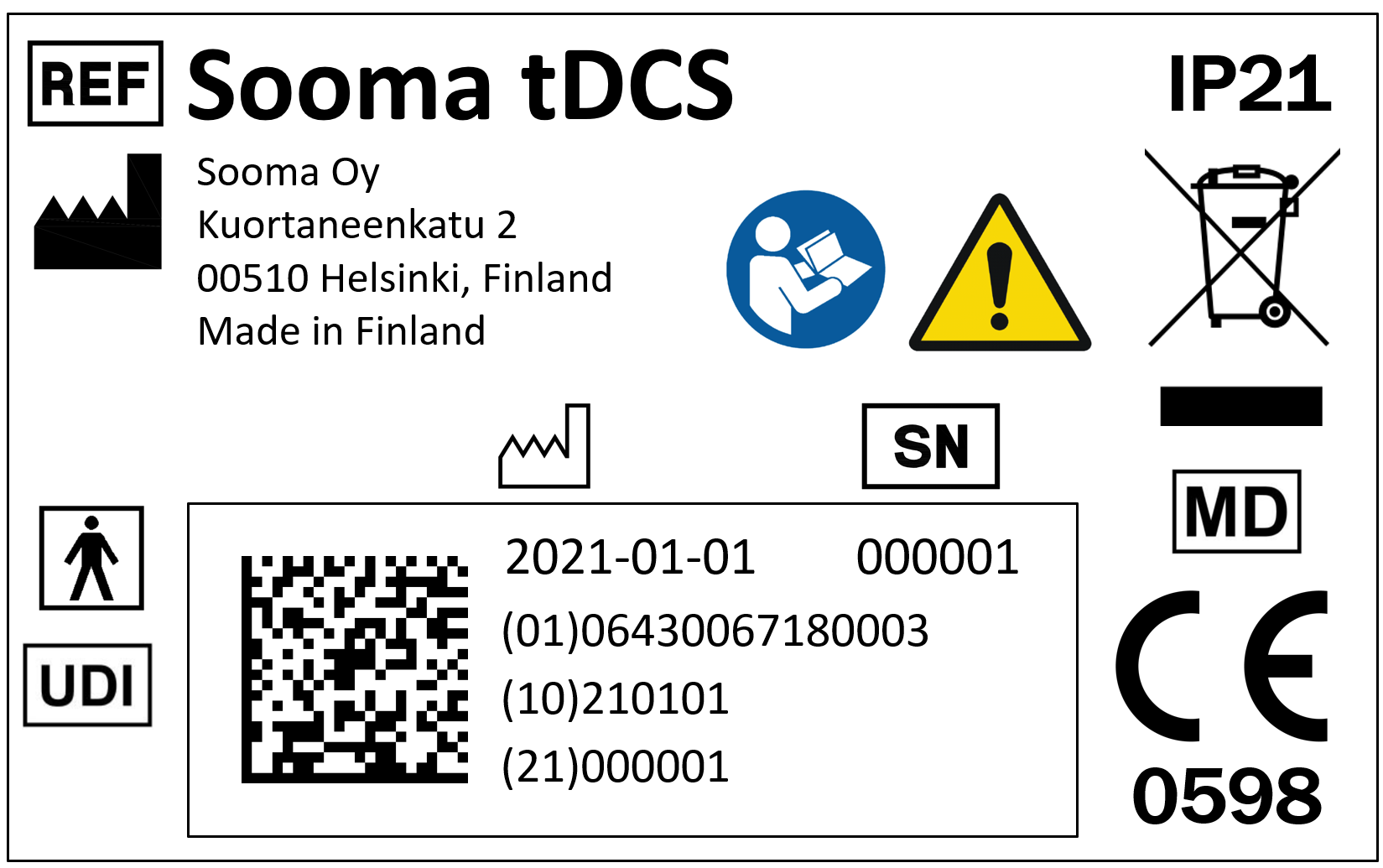
There is also a standard that regulates the symbols to be included in CE produced devices. In a few years, such standard will include a “medical device” symbol (market by the letters MD), so in the future that symbol can be sought to corroborate that the product is in fact a medical device.
Latest news

Sooma and Clariane launch multi-country pilot following successful EIC Corporate Partnership Programme
Read more
Active, not just treated: Dr. Ulrike Vogelmann on Brain Stimulation for Depression
Read more
Treating Depression Differently: Integrating tDCS Into Your Treatment Pathway
Read more